rpsL DNA sequence analysis protocol
1. Download and install ApE: ApE website
Installing ApE might be the most challenging part!
On a Mac:
Download and open ApE dmg file.
Drag ApE.app into Applications.
Follow the install directions on the ApE webpage.
If that does not work, try this:
Go to Applications --> Utilities --> Terminal.app
Double-click Terminal.app to launch
In the window that pops up, type in (or copy/paste) and hit return:
xattr -d com.apple.quarantine /Applications/ApE.app
Close the window to quit Terminal
Go back to Applications.
Double-click ApE.app to start program
2. Go to rpsL DNA sequence data and download the file that matches your sequence tube code.
3. Open the .ab1 file with ApE.
1. Download and install ApE: ApE website
Installing ApE might be the most challenging part!
On a Mac:
Download and open ApE dmg file.
Drag ApE.app into Applications.
Follow the install directions on the ApE webpage.
If that does not work, try this:
Go to Applications --> Utilities --> Terminal.app
Double-click Terminal.app to launch
In the window that pops up, type in (or copy/paste) and hit return:
xattr -d com.apple.quarantine /Applications/ApE.app
Close the window to quit Terminal
Go back to Applications.
Double-click ApE.app to start program
2. Go to rpsL DNA sequence data and download the file that matches your sequence tube code.
3. Open the .ab1 file with ApE.
4. Download the rpsL_PCR.xdna file using "save link as file" option. Open it with ApE. In this file, the rpsL START codon and STOP codon are in LOWERCASE.
5. Go to Tools -> "Align Sequences"
6. In the window that comes up, select the top “window” to your .abi trace file, and the other to rpsL_PCR, and click OK. Leave the other settings at the default values.
7. The alignment window should show your sequence on top (your sequence) aligned to the wild-type on the bottom. Mis-matches, including gaps, will be highlighted. Ignore all except those between the start codon and stop codon; the sequence is of poor quality at the start and end of a read.
8. Double-click on a base on the top sequence in the alignment window. This will take you to that base in the chromatogram. Confirm that the chromatogram matches the called sequence by your eye; ie: make sure the computer didn’t make a mistake! Repeat for all the differences, and note changes (base position and what you think it should be).
9. Close the alignment window.
10. Go to the .abi window. Use File -> "New DNA from basecalls". This opens a window with the chromatogram sequence ("Q5.abi" gets converted to "Q5.ab1 basecalls" for example). Close the .abi trace file
11. Go to Tools -> "Align Sequences". In the window that comes up, select the top “window” to your basecalls file, and the other to rpsL_PCR, and click OK. Leave the other settings at the default values.
12. The alignment window should show your sequence on top (your sequence) aligned to the wild-type on the bottom. Find the correct start codon in your sequence. In the alignment window, double-click on a base above the wild-type gene's start codon (lower-case). This will bring you to the basecalls window. Make the start codon lowercase by highlighting the codon and Edit -> "convert to lowercase".
13. Repeat step 12 for the stop codon, which will also be in lower-case in the wild-type sequence.
14. Mis-matches, including gaps, will be highlighted. Ignore all except those between the start codon and stop codon; the sequence is of poor quality at the start and end of a read. Double-click on a base on the top sequence in the alignment window. This will take you to that base in the basecalls file. Convert it to lowercase by highlighting the base and Edit -> "convert to lowercase". Do the same to the wild-type sequence positions that differ from your mutant. After this step, both the wild-type and basecalls files will have the start codon, stop codon, and differing bases in lower-case.
15. Go the the basecalls window. Go to Edit -> "UPPER<->lower". This will now make the lower-case letters upper-case and vice-versa. Do the same for the wild-type file.
16. Delete the bases before and after the start and stop codon in your basecalls file. The total coding sequence length, including start and stop codons is 375 bases. Save the basecalls file.
17. Repeat step 16 for the wild-type file. Rename and save the trimmed wild-type file.
5. Go to Tools -> "Align Sequences"
6. In the window that comes up, select the top “window” to your .abi trace file, and the other to rpsL_PCR, and click OK. Leave the other settings at the default values.
7. The alignment window should show your sequence on top (your sequence) aligned to the wild-type on the bottom. Mis-matches, including gaps, will be highlighted. Ignore all except those between the start codon and stop codon; the sequence is of poor quality at the start and end of a read.
8. Double-click on a base on the top sequence in the alignment window. This will take you to that base in the chromatogram. Confirm that the chromatogram matches the called sequence by your eye; ie: make sure the computer didn’t make a mistake! Repeat for all the differences, and note changes (base position and what you think it should be).
9. Close the alignment window.
10. Go to the .abi window. Use File -> "New DNA from basecalls". This opens a window with the chromatogram sequence ("Q5.abi" gets converted to "Q5.ab1 basecalls" for example). Close the .abi trace file
11. Go to Tools -> "Align Sequences". In the window that comes up, select the top “window” to your basecalls file, and the other to rpsL_PCR, and click OK. Leave the other settings at the default values.
12. The alignment window should show your sequence on top (your sequence) aligned to the wild-type on the bottom. Find the correct start codon in your sequence. In the alignment window, double-click on a base above the wild-type gene's start codon (lower-case). This will bring you to the basecalls window. Make the start codon lowercase by highlighting the codon and Edit -> "convert to lowercase".
13. Repeat step 12 for the stop codon, which will also be in lower-case in the wild-type sequence.
14. Mis-matches, including gaps, will be highlighted. Ignore all except those between the start codon and stop codon; the sequence is of poor quality at the start and end of a read. Double-click on a base on the top sequence in the alignment window. This will take you to that base in the basecalls file. Convert it to lowercase by highlighting the base and Edit -> "convert to lowercase". Do the same to the wild-type sequence positions that differ from your mutant. After this step, both the wild-type and basecalls files will have the start codon, stop codon, and differing bases in lower-case.
15. Go the the basecalls window. Go to Edit -> "UPPER<->lower". This will now make the lower-case letters upper-case and vice-versa. Do the same for the wild-type file.
16. Delete the bases before and after the start and stop codon in your basecalls file. The total coding sequence length, including start and stop codons is 375 bases. Save the basecalls file.
17. Repeat step 16 for the wild-type file. Rename and save the trimmed wild-type file.
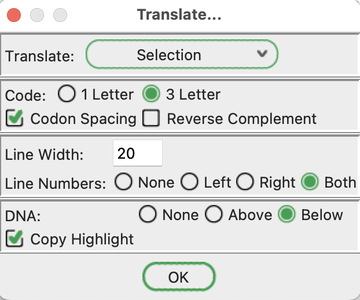
18. Go to the basecalls window. Go to ORFs -> Translate.
In the window that pops up:
Select "3 letter" for "Code".
Select "Codon Spacing".
Enter "20" for "Line Width".
Select "Both" for "Line Numbers".
Select "Below" for "DNA".
Select "Copy Highlight".
Click OK.
This will create a new window containing the amino acid sequence above each codon.
19. Repeat step 18 for the wild-type sequence.
20. Put the two translation windows (wild-type, your sequence) side-by-side. Look for capital letters in the DNA (bottom) ... those positions are mutated in your sequence.
What positions are mutated at the DNA level (DNA base numbers at the start and end of each row)?
What amino acid position do these mutations correspond to (amino acid base numbers at the start and end of each row)?
What is the wild-type base, codon, and amino acid at that position?
What is the mutant base, codon, and amino acid at that position?
If the amino acid is the same, how would you classify this mutation?
If the amino acid is different between wild-type and mutant, is this mutation conservative (same class of amino acid; ie: hydrophobic --> hydrophobic, negative --> negative, etc)? Or is it non-conservative (ie: hydrophobic --> positive, etc)?
In the window that pops up:
Select "3 letter" for "Code".
Select "Codon Spacing".
Enter "20" for "Line Width".
Select "Both" for "Line Numbers".
Select "Below" for "DNA".
Select "Copy Highlight".
Click OK.
This will create a new window containing the amino acid sequence above each codon.
19. Repeat step 18 for the wild-type sequence.
20. Put the two translation windows (wild-type, your sequence) side-by-side. Look for capital letters in the DNA (bottom) ... those positions are mutated in your sequence.
What positions are mutated at the DNA level (DNA base numbers at the start and end of each row)?
What amino acid position do these mutations correspond to (amino acid base numbers at the start and end of each row)?
What is the wild-type base, codon, and amino acid at that position?
What is the mutant base, codon, and amino acid at that position?
If the amino acid is the same, how would you classify this mutation?
If the amino acid is different between wild-type and mutant, is this mutation conservative (same class of amino acid; ie: hydrophobic --> hydrophobic, negative --> negative, etc)? Or is it non-conservative (ie: hydrophobic --> positive, etc)?